
Next generation sequencing (NGS) methods allow rapid and high throughput sequencing of large stretches of DNA base pairs, which can be used for direct detection of mutations without any pre-screening. It is possible to do simultaneous screening of several genes for identification of allelic variants.
AgriGenome can offer services coupled with other techniques like CRISPR.

TILLING (Targeting Induced Local Lesions IN Genomes) is a reverse genetic technique that uses traditional chemical mutagenesis methods to create libraries of mutagenized individuals that are later subjected to high-throughput screens for the discovery of mutations. Previous TILLING protocols employed enzymatic or physical discrimination of heteroduplexed from homoduplexed target DNA. But these methods are very laborious, time-consuming, need extensive automation and sometimes have lower sensitivity and resolution than needed and the heteroduplexes need to be confirmed by Sequencing. AgriGenome offers mutation screening method based on Illumina sequencing of target genes amplified from multi-dimensionally pooled templates representing 768 individuals per experiment.
AgriGenome, in collaboration with SciGenom, has taken the initiative to catalogue the variant information of various rice varieties using its analysis pipeline. RiceVarDb is a database of variants identified from ~ 80 different varieties of rice. The raw reads retrieved from various public data repositories like short-read archives have been analyzed using a standard variant calling pipeline against the Nipponbare MSU6 genome assembly and the variants have been recorded in the database. Variant annotation analysis has been performed using various in-house programs and databases. The database can be searched using gene and transcript identifiers. RiceVarDb provides overall allele information, across the rice lines, along with the gene structure and annotation. RiceVarDb presents allelic information that are organized into six different groups, namely Indica, Temperate Japonica, Tropical Japonica, Aromatic, Aus and Wild lines, with an appropriate graphical view to enable a comparative visualization of the variants and their contributions. RiceVarDb is expected to serve as a highly informative visualization tool for allele mining and exploring allele sharing patterns between selected lines at the target loci for molecular mapping and marker assisted selection.
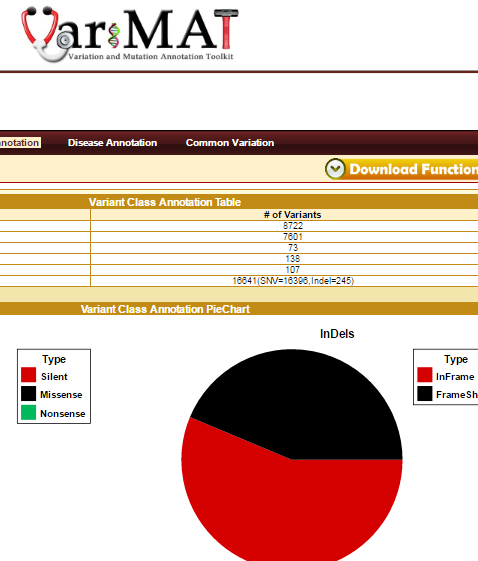
VariMAT is a web-based toolkit for comprehensive annotation of human single nucleotide variations (SNVs) and shot insertions/deletions (indels). VariMAT utilizes different databases and public resources including UCSC, GENCODE, ENCODE, Ensembl, dbSNP, NCBI, HapMap, and the 1000-genomes project. The various annotation types implemented in the toolkit are: genomic, functional, common variation and disease annotation. Genomic annotation includes genes, splice-sites, UTRs, repeats, conserved transcription factor binding sites (TFBS), regulatory regions (from ChiP-seq), conserved enhancers, CpG-Islands, miRNAs and promoter regions. The functional annotations implemented into the toolkit are variant class prediction (silent, missense, nonsense, inframe and framshift), polyPhen and gene ontology summary. Various disease annotation includes are OncoMD, GWAS, ClinVar, SNPedia. In addition, the tool also compares the variant with several published personal genomes and common variation databases – dbSNP, 1000-genomes, HapMap. It accepts input in variant call format (vcf) and 23andMe format. VariMAT is capable of annotating several thousand variants in minutes. The complete annotation result can be easily exported to the local desktop computer. It also generates several tables and plots that can be used for publications.

Designing primer for experimental validation is one most frequent task performed by a biologist. Primer designing involve several steps and is a time consuming procedure. PrimeMe is a web-based toolkit to simply and speed up the primer designing process. PrimerMe provides various search options including i) search by chromosome ii) search by gene/transcript iii) search by SNP. One can also upload a list of coordinates to design primer. The current version of PrimeMe support hg19 version of the human genome and use RefSeq gene model to identify gene and transcript position. It also includes 5.5 million SNP coordinates from snp135 database.User can design primer for either complete gene region or exonic regions. While deigning primer user can set primer condition and primer type – PCR or Primer walking. The result for primer design is displayed in both graphical and tabular format with SNP being highlighted in the primer sequences if any. A link to the UCSC Insilco PCR is provided to find the number of products the primer set would provide. One could also filter out the primers if a SNP is present in the first 3 bases in the primer 5' end. The complete result can be downloaded into the user's local system in a text file. Results can also be saved in the PrimeMe database for future use.
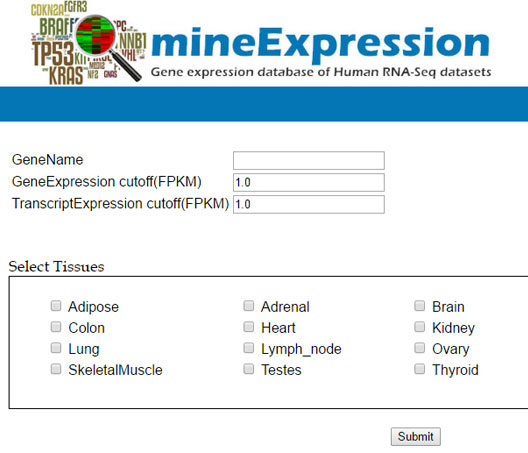
mineExpression is a database of gene and transcript expression for various tissue and cell lines. The RNA-seq datasets are downloaded from various public databases and are analyzed using custom RNA-seq pipeline at AgriGenome. The expression value estimated using our in house pipeline is deposited into mineExpression database. A gene-based search is provided to extract expression value at gene and transcript level. User can select a cut-off value while searching and can also select one or more tissues/cell lines.

Mutations in the mitochondrial genome are one of the significant reasons for various inherited diseases in humans. Mitochondrial genome size is only 16Kb and can easily be sequenced by next-generation sequencers at low cost and high depth. Mutation identified in the mitochondrial genome using the NGS technique need to be annotated for previously reported disease mutations. A gene and region based search is provided in MitoGIS to look for disease related mutations. MitoGIS can also annotate mutations identified using NGS approach. It accepts input as vcf file format.
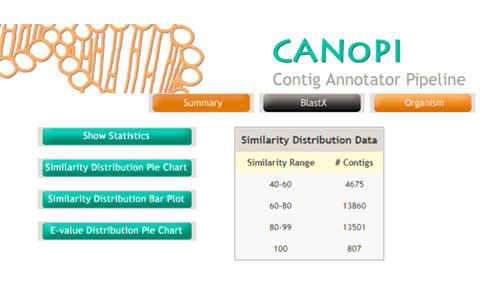
Denovo genome and transcriptome assembly generates several thousands of contigs. It is very important to perform comprehensive annotation of predicted genes and transcripts. The CANoPI annotation pipeline is design to automatic annotate the contig with various databases. CANoPI results include BLASTX search result, similarity with known proteins at NCBI, provides gene ontology (GO) summary, UniProt annotation and species overlap summary. The pipeline generates several statistical charts and tables required for publications.
Analysis of next-generation sequencing (NGS) data set is a huge challenge. It needs a systematic and intelligent approach to process the NGS data efficiently. AgriGenome has developed workflows and programs to analyze large-scale biological data sets, especially focused towards NGS. Our analysis process includes data quality assessment, comprehensive analysis, interpreting results, and communicating and presenting results to the customers in meaningful formats.